1.1. DEFINITION
Idiopatisk pulmonal fibrose (IPF) er en specifik form for kronisk progredierende fibrotisk interstitiel lungesygdom (ILS) af ukendt ætiologi. IPF er udelukkende lokaliseret i lungerne og ses kun hos voksne.
Det dominante træk ved IPF er irreversibel udvikling af lungefibrose med et radiologisk-patologisk mønster kaldet usual interstitial pneumonia (UIP). Diagnosen kræver tilstedeværelse af 1 samtidig med 2 og/eller 3.
- Eksklusion af anden idiopatisk interstitiel lungesygdom eller anden interstitiel lungesygdom med kendt årsag til UIP og
- UIP-mønster på HRCT og/eller
- Patologisk UIP mønster
IPF repræsenterer en specifik klinisk sygdomsentitet, mens UIP er et radiologisk-patologisk mønster, der også kan ses ved andre former for ILS, som fibrotisk allergisk alveolitis og reumatologiske sygdomme med lungeinvolvering som f.eks. reumatoid artritis og systemisk sklerodermi.
1.2. FOREKOMST
Der findes ingen eksakte danske tal for incidens og prævalens af IPF, men med en estimeret incidens på ca. 400 patienter pr. år og en prævalens på ca. 6-7/100.000. Såvel incidens som prævalens stiger med alderen.
1.3. SYMPTOMER OG KLINISKE FUND
IPF optræder kun hos voksne og oftest hos personer over 50 år og med overvægt af mænd (67%) og rygere/eksrygere (75%). I differentialdiagnostisk øjemed bør man altid mistænke og undersøge for underliggende reumatologisk sygdom. Typisk ses snigende udvikling af funktionsdyspnø og tør hoste over få måneder til år. Objektivt høres typisk tør fibroseknitren (velcrokrepitation) basalt ved lungestetokopi, og ofte ses trommestikfingre og urglasnegle (clubbing).
1.4. UDREDNING OG DIAGNOSE
Udredningen af IPF følger de gængse retningslinjer for udredning af interstitielle lungesygdomme (Se DLS retningslinjen ”Interstitielle lungesygdomme”). Det er vigtigt at udskille IPF fra andre typer af ILS, idet IPF-diagnosen har stor betydning for prognose og valg af behandling.
Udredningen fokuserer på:
- Grundig anamnese med vurdering af, om der kan identificeres relevant eksponering, der kan udløse ILS.
- HRCT med vurdering af tilstedeværelse af UIP-mønster.
- Multidisciplinær team (MDT) konference. Her tages stilling til om klinik og HRCT er tilstrækkelig til at stille diagnosen IPF eller om der er behov for:
- Bronkoskopi med BAL til celletælling og lungebiopsi (cryobiopsi) til histopatologi alternativt kirurgisk biopsi.
- Herefter ny MDT konference
1.4.1. Anamnese:
For at stille diagnosen IPF er det væsentligt at udelukke andre kendte årsager til lungefibrose som f.eks. medikamentel-, erhvervsmæssig-, eller miljøudløst eksponering, men også underliggende autoimmune reumatologiske sygdomme. Dette sikres ved en grundig anamnese, evt. understøttet af specifikke patientudfyldte spørgeskemaer. Der skal i anamnesen lægges vægt på sygdomsudvikling, alder, køn og rygeanamnese, tilstedeværelse af tegn og symptomer på reumatologisk sygdom, anden systemsygdom med evt. lungeinvolvering, samt eksponering, der kan medføre allergisk alveolitis (Se DLS retningslinje om ”Allergisk alveolitis”). Derfor også grundig anamnese omkring erhverv, boligforhold, hobbies, medikamenter og arvelighed. Ved mistanke om erhvervsmæssig eller reumatologisk årsag bør patienten henvises til vurdering ved de respektive specialer. Mhp. differentialdiagnostisk udelukkelse eller understøttelse af mistanke om systemsygdom foretages måling af autoimmune blodprøver.
1.4.2 HRCT
Det radiologiske mønster inddeles i fire grupper: ”UIP-mønster”, ”Probable UIP-mønster”, ”Indeterminate UIP-mønster” og “HRCT-fund tydende på en alternativ diagnose” (se nedenfor).
UIP-mønster (alle kriterier opfyldte):
- Overvejende subpleural, basal lokalisering
- Lokalisering er ofte heterogen
- Nogle gange diffust eller asymmetrisk (op imod 25%)
- Bikagetegning med eller uden traktionsbronkiektasier eller traktionsbronkiolektasier*
- Irregulær fortykkelse af intra- og interlobulære septae (retikulært mønster)
- Confidence-grad for et histopatologisk UIP-mønster hos > 90%
Probable UIP-mønster (alle kriterier opfyldte):
- Subpleural, basal beliggenhed
- Lokalisering er ofte heterogen
- Retikulært mønster med perifere traktionsbronkiektasier eller traktionsbronkiolektasier
- Der kan være let matglastegning i områder med retikulering*
- Ingen subpleural sparring
- Confidence-grad for et histopatologisk UIP-mønster hos 70-89%
Indeterminate UIP mønster:
- Diffus lokalisering uden subpleural beliggenhed
- Tegn på fibrose, der ikke tyder på et specifikt mønster
- Confidence-grad for et histopatologisk UIP-mønster hos 50-69%
CT-fund tydende på en alternativ diagnose:
Beliggenhed:
- Øvre eller midt lunge prædominans
- Peribronkovaskulær dominans med subpleural sparing
- Perilymfatisk beliggenhed
- Subpleural sparing
Specifikke fund:
- Cyster
- Mosaik attenuering
- Konsoliderede infiltrater
- Udbredt matglasforandringer (hvis ikke akut exacerbation)
- Udbredte centrilobulære mikronoduli
- Noduli
- Pleurale plaques
- Dilateret øsofagus
- Histopatologisk UIP-mønster hos < 50%
*Retikulært mønster beliggende oveni matglasforandringer er oftest tegn på fibrose, især hvis der samtidig ses traktionsbronkiektasier. Rene matglasforandringer taler imod UIP/IPF og vil antyde akut exacerbation, allergisk alveolitis eller andre ILS tilstande.
1.4.3. Histopatologi
Det histopatologiske mønster beskrives som ”UIP-mønster”, ”Probable UIP mønster”, ”Indeterminate UIP mønster” og “Bedst foreneligt med alternativ diagnose” (se nedenfor).
UIP-mønster (alle kriterier opfyldte):
- Udtalt fibrose/arkitektonisk distortion
- +/- bikagetegning overvejende subpleuralt/ paraseptalt
- Tilstedeværelse af heterogenicitet
- Tilstedeværelse af fibroblast foci
- Fravær af fund tydende mod UIP eller tydende på anden diagnose
Probable UIP-mønster:
- En kombination af ovenstående fund i en grad, der ikke kan stille en sikker UIP diagnose og
- Fravær af fund tydende mod UIP eller tydende på anden diagnose eller
- Bikagetegning alene
Indeterminate UIP-mønster (alle kriterier opfyldte):
- Tegn på fibrose med træk der mere peger i retning af non-UIP eller non-IPF UIP
- Mindre overbevisende UIP-fund og evt. tegn på:
- Foci med centrilobulær affektion
- Enkelte granulomer eller kæmpeceller
- Forekomst af centrilobulær eller lymfoid hyperplasi
- Diffus inflammation
- Diffus homogen fibrose tydende på NSIP
Mest tydende på alternativ diagnose (tilstedeværelse af et hvilket som helst kriterie):
- Fibrotisk allergisk alveolitis
- Fibrotisk NSIP
- Hyaline membraner (kan være associeret med akut exacerbation af IPF)
- Fibrotisk organiserende pneumoni (FOP)
- Pleuroparenkymatøs fibroelastose (PPFE)
- Pulmonal Langerhans celle histicytose (PLCH)
- Ryge-relateret interstitiel fibrose
- UIP med ledsagende fund, der antyder anden diagnose som f.eks.
- Diffus alveolær damage (DAD)
- Organiserende pneumoni (OP)
- Granulomer
Udtalt interstitielle inflammatoriske celle infiltrater uden relation til UIP
1.4.4. Multidisciplinære team (MDT) konference
IPF-diagnosen anbefales opnået ved MDT konference med deltagelse af lungemediciner, radiolog og evt. patolog, reumatolog og arbejdsmediciner. Sikkerheden af IPF-diagnosen afhænger af det radiologiske mønster, og om der evt. foreligger histopatologi. Når de klassiske HRCT-forandringer er forenelige med ”UIP-mønster” eller ”probable UIP-mønster” og er til stede i den rette kliniske kontekst, er den histopatologiske diagnose med høj sandsynlighed UIP, og der er derfor ikke behov for biopsi. Ved ”probable UIP-mønster” kan BAL overvejes, hvis den kliniske kontekst giver mistanke om kronisk allergisk alveolitis eller fibrotisk NSIP. Lungebiopsi og BAL anbefales, hvor den kliniske kontekst ligner IPF, men HRCT er vurderet som ”indeterminate UIP-mønster” eller ”mest forenelig med alternativ diagnose”, eller hvor den kliniske kontekst ikke er som ved IPF. Hos de patienter, hvor biopsi findes kontraindiceret pga. sygdommens sværhedsgrad, komorbiditeter eller manglende samtykke fra patienten, må man stille den mest sandsynlige diagnose (arbejdsdiagnose) og notere om diagnosen er stillet med høj (90-100%), moderat (70-89%) eller lav (50-69%) sikkerhed. Graden af diagnostisk sikkerhed dokumenteres i patientens journal med evt. samtidig plan for, hvornår diagnosen skal revurderes. Ved MDT konference tages også stilling til behandlingstilbud.
1.5. BEHANDLING
Der findes ingen kurativ behandling af IPF, men antifibrotisk behandling er i flere studier vist at nedsætte sygdomsprogression via et reduceret årligt fald i FVC, forlængelse af tid til akut exacerbation (AE-IPF) og indlæggelse, som samlet således medfører en reduceret mortalitet. Ubehandlet vil patienter med IPF gennemsnitligt miste ca. 200 ml i FVC årligt uanset sværhedsgrad af sygdommen. Det er derfor væsentligt at henvise patienten tidligt i sygdomsforløbet mhp. tidlig initiering af antifibrotisk behandling.
1.5.1. Medikamentel behandling af IPF
Pirfenidon og nintedanib er begge antifibrosemidler, som i undersøgelser er vist at besidde ovennævnte egenskaber. Indikationen er mild til moderat IPF (DLCO > 30% og FVC > 50%), men real-life studier viser tilsvarende behandlingseffekt hos patienter med avanceret IPF. Antifibrotisk behandling er en specialistbehandling og varetages på de fire ILS centre (Herlev-Gentofte Hospital, Rigshospitalet, Odense Universitetshospital og Aarhus Universitetshospital).
Et behandlingsrespons vil vise sig som uforandret status eller reduceret progressionshastighed. Patienten bør derfor informeres om, at behandlingen alene forventes at mindske progression og ikke bedrer lungefunktionen.
Bivirkninger:
Behandling med både pirfenidon- og nintedanib kan være forbundet med en del bivirkninger. Særligt gastrointestinale bivirkninger er hyppige. Bivirkningerne forsøges altid afhjulpet, primært med understøttende behandling, og først herefter dosisreduktion, da der er dosis-respons effekt. Behandling og monitorering varetages af de fire ILS centre.
Pirfenidon:
Typisk nedsat appetit, madlede og evt. kvalme/opkastninger førende hos nogle til et større vægttab. Bør understøttes med anti-emetika og kosttilskud.
Udslæt evt. i form af fototoxisk eksantem ses hos knap 20%, hvorfor patienterne anbefales solcreme med faktor 50 dækkende både UVA og UVB-stråler, tildækning af soleksponerede overflader (hat, langærmede skjorter m.m.) samt at undgå direkte soleksponering.
Leverpåvirkning: 2-3% får transaminasæmi, hvorfor levertal kontrolleres jævnligt og behandlingen seponeres, hvis ALAT er forhøjet > x 3.
Nintedanib:
Typisk nedsat appetit, madlede og evt. kvalme/ opkastninger førende til et større vægttab hos nogle patienter. Bør understøttes med anti-emetika og kosttilskud.
Diarre ses hos op mod 70% i varierende grad og kan hos enkelte være invaliderende og føre til behandlingsophør. Diarre behandles med væsketilskud og loperamid, enten fast eller p.n.
Leverpåvirkning: 2-3% får transaminasæmi, hvorfor levertal kontrolleres jævnligt og behandlingen seponeres, hvis ALAT er forhøjet > x 3.
Generelt gælder det om at fastholde patienterne i maksimal behandling ved understøttende tiltag før dosisreduktion, da effekten af antifibrotisk behandling, herunder livsforlængelse, er dosisafhængig. Patientens præferencer skal naturligvis inddrage i behandlingsbeslutninger.
Ophør med behandling:
Antifibrotisk behandling fortsættes så længe den tolereres, og så længe det er meningsfuldt. Det gælder også under akutte indlæggelser med exacerbation samt ved faldende FVC og DLCO. Kun ved uacceptable og behandlingsresistente bivirkninger bør der ske ophør eller skift af behandling.
- Understøttende og palliativ behandling
- Iltbehandling påbegyndes efter vanlige kriterier. Ambulatorie oxygen terapi (AOT, ”funktionsilt”) kan tilbydes ved desaturation < 88% med heraf demonstreret effekt i form af reduceret desaturation, længere gangdistance og/eller sikker subjektiv forbedring efter 3 måneders behandling (Se DLS retningslinjen ”Iltbehandling”).
- Protonpumpeinhibitorer gives ved symptomer på GERD.
- Morfin og kodein gives mod åndenød, angst og hoste.
- Lungerehabilitering bør tilbydes alle mobile patienter.
- Patienter med pulmonal hypertension har dårlig prognose. Treprostinil, en prostacyclinanalog, er i et enkelt studie vist at bedre funktionsniveau målt ved 6-minutters gangtest, men er ikke godkendt til behandling af ILS-relateret PH (WHO grp. III) i Danmark.
1.5.2.1. Prednisolonbehandling
Hos patienter med svær sygdom kan lavdosis prednisolon forsøges i palliativt øjemed mhp. bedring af bl.a. almentilstand, appetit og vægt samt reduktion af hoste. Højdosis prednisolon har ingen sygdomsmodificerende effekt og bør undgås, da der er set forøget risiko for infektion, indlæggelse og død.
1.5.2.2. Terminalstadiet
Mange patienter har i terminalstadiet glæde af tilknytning til de lokale palliative teams, som bl.a. åbner op for vederlagsfri medicin, hjælpemidler, fysioterapi, psykologibistand, og plejeorlov til pårørende. I forhold til sidstnævnte kræves separat ansøgning om terminaltilskud på FMK-online.dk. Der bør laves terminalerklæring, når sygdommen er tilstrækkelig fremskreden (se DLS’ ”Klaringsrapport om palliation til nonmalign kronisk lungesygdom”).
1.5.3. Lungetransplantation
Patienter uden tegn på svær komorbiditet bør tidligt vurderes med henblik på muligheden for lungetransplantation. Henvisning til udredning bør overvejes, når DLCO < 40%, behov for iltbehandling inkl. AOT, fald i FVC (>10%) og/eller DLCO (>15%) over de seneste 2 år, fald i FVC og/eller DLCO sammen med tiltagende symptomer og/eller tiltagende fibrose på HRCT. Se DLS-retningslinjen om ”Lungetransplantation”.
1.6. OPFØLGNING
Patienterne bør følges jævnligt, oftest hver 3. – 6. måned, med vurdering af bl.a. lungefunktion, 6 minutters gangtest, blodprøver ved antifibrotisk behandling, saturation og behov for palliation m.m.
1.7. AKUTTE EXACERBATIONER VED IPF (AE-IPF)
AE-IPF er defineret som ”Akut signifikant klinisk respiratorisk forværring karakteriseret ved ny og udbredt alveoleskade”. Se DLS retningslinjen ”Akut exacerbation af IPF og andre interstitielle lungesygdomme”.
Antifibrotisk behandling skal ikke pauseres under en AE-IPF, da antifibrotika ikke øger risikoen for infektion m.m. Et enkelt studie har vist, at start af antifibrotisk behandling under AE-IPF hos patienter uden tidligere kendt IPF forlænger overlevelsen.
1.8. FOREBYGGENDE BEHANDLING
Som ved andre kroniske lungesygdomme anbefales patienterne tobaksophør og vaccination for pneumokokker, influenza og Covid-19.
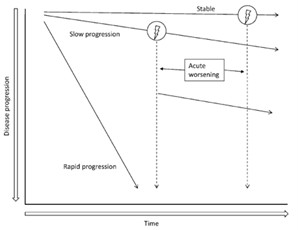
1.9. PROGNOSE
IPF er en uundgåelig progredierende fibrotisk lungesygdom med en medianoverlevelse på 3-5 år når ubehandlet. Observationelle studier efter indførelse af antifibrotisk behandling indikerer en forlænget medianoverlevelse på mere end 5 år.
Den enkelte patients sygdomsforløb kan være uforudsigeligt med stabile faser og faser med fald i lungefunktion. De fleste patienter oplever progression over få år. Nogle patienter kan være stabile i en årrække, andre oplever ganske hurtig progression over få måneder, evt. i form af AE-IPF.
GAP score (Gender, Age, Physiology (FVC og DLCO)) er en valid prædiktor for død, men ikke for sygdomsprogression.
1.10. REFERENCER
Raghu G et al; Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. AJRCCM 2018 Sep 1;198(5): e44-e68
Raghu G et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med 2022; 205(9): e18-e47.
King et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonafry fibrosis. NEJM 2014;370;2083-92.
Richeldi et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. NEJM 2014; 370: 2071-80.
Raghu et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. AJRCCM 2015; 192: e3-e19.