
1.1. DEFINITION
Allergisk alveolitis (AA) eller hypersensitivitets pneumonitis (HP) er en inflammatorisk og/eller fibrotisk sygdom, der afficerer lungeparenkymet og de små luftveje. AA skyldes typisk en im munmedieret reaktion provokeret ved inhalation af et kendt eller okkult antigen hos modtagelige individer.
1.2. FOREKOMST
Et dansk registerstudie har fundet en AA inci-dens på 1,16/100.000 personer. Hyppigheden af AA er stigende og i et amerikansk studie fandt man en estimeret 1-års prævalens på 1,67 stigende til 2,71/100.000 indbyggere fra 2004 til 2015. Stigningen sås især > 65 år, hvor prævalensen var 11/100.000. Over 300 forskellige antigener er identificeret som årsag til AA, hvoraf de fleste er case re- ports, enkeltudbrud og flere nu kun af historisk interesse.
1.3. PATOGENESE
AA repræsenterer en immunologisk reaktion på et inhaleret antigen. Patogenesen er kompleks og kun delvist belyst, men involverer både im munkompleks medieret (type III) og T-celle medieret (type IV) reaktioner hos genetisk di sponerede personer. Immunkompleks reaktio nen har den betydning at serum præcipiterende antistoffer mod det udløsende antigen kan de monstreres i blodet.
AA dækker over mange forskellige eksponerin ger og kliniske præsentationer. Forekomsten varierer betydeligt mellem klimazoner, erhverv og arbejdsmetoder, f.eks. i landbruget. Patienter med AA har været udsat for gentagen inhalation af aerosoler/støv som f.eks. fuglestøv, dampe eller gasser, hvori de egentlige antigener befin der sig. Dette kan være bakterier, svampe, pa rasitter, dyre- eller plante-proteinstoffer, typisk organiske, men nogle gange uorganiske kemika lier.
De hyppigste og bedst undersøgte former for AA er fugleholderlunge, hvis årsag både kan skyl des fækale og fjerrelaterede proteiner. Tidligere var tærskerlunge hyppig, hvor bakterien Saccha ropolyspora rectivirgula er en af de vigtigste patogener inden for Thermophilic actinomycetes slægten, men med ændrede landbrugsmetoder er forekomsten faldet betydeligt.
AA kan skyldes skimmelsvampeantigener fra fugtigt træ f.eks. hos savværksarbejdere, arbej de med flis til opvarmning, skimmelvækst i bl.a.

Rygning eller virkningerne heraf har betydning for forekomsten af AA, idet AA oftere ses hos ikke-rygere end hos rygere, mens rygere med AA ofte har et alvorligere forløb end ikke-rygere med AA.
1.4. UDREDNING
AA klassificeres klinisk efter tilstedeværelse af inflammation og/eller fibrose på HRCT og/eller histopatologi som non-fibrotisk eller fibrotisk AA. Patienter med blandet morfologi karakteriseres baseret på den dominerende morfologi. Fore
komst af fibrose er dog oftest bestemmende for behandlingsrespons og prognose.
Fibrotisk AA kan være meget svær at skelne fra andre former for fibrotisk interstitiel lungesyg dom. Det kliniske billede er broget, men detalje ret anamnese med fokus på eksponering, symp tomatologi, objektive fund inklusiv diverse parak linik, samt multidisciplinær team diskussion med tilstedeværelse af lungemedicinere, radiologer, patologer og arbejdsmedicinere, kan føre til mere præcis diagnostik.
Eksponering
Meget væsentligt er nøje oplysning om ekspone ring (f.eks. hønsehold, duer, volierefugle, land brugspartikler, foderstoffer, træfældning, vand skade m.m.), udluftningsforhold, synlig
fugt/skimmelsvamp og opmærksomhed på evt. medicin, ligesom opmærksomhed på varighed og debut af symptomer også er væsentlig i rela tion til eksponering.
Symptomer
Der er stor symptomvariation ved non-fibrotisk og fibrotisk AA afhængig af intensitet og fre kvens af eksponering for det årsagsgivende antigen. Der er et betydeligt overlap mellem fænotyperne.
Non-fibrotisk AA
Er typisk karakteriseret ved debut timer til få dage efter korttids/intermitterende eksponering med udvikling af feber, hoste, dyspnø, og vedva rende (uger) influenzalignende sygdomsfor nemmelse. Symptomerne kan bedres væsentligt mellem eksponeringsperioderne. Ved fortsat eksponering kan hosten blive produktiv, ånde nøden permanent og almentilstanden perma nent nedsat.
Fibrotisk AA
Er typisk karakteriseret ved længere (måneder år) sygdomsvarighed. Fremtrædende sympto mer er ofte hoste, snigende dyspnø, træthed og vægttab. Rapportering af tidligere akutte og subakutte episoder vil ofte være en hjælp til at identificere tilstanden, men kan helt mangle i sygehistorien. Akut exacerbation af fibrotisk AA kan ses og har et alvorligt forløb med høj morta litet (se DLS retningslinje ”Akut exacerbation af IPF og andre interstitielle lungesygdomme”).
1.4.1 Objektive fund
Objektive fund er sparsomme og uspecifikke med takypnø og tør, fin krepitation eller sjældne re inspiratoriske rhonchi (eng. squeaks) ved lungestetoskopi. Ved svær AA ses hyppigt desa turation ved aktivitet eller hypoxæmi uden at dette fund er specifikt for selve sygdommen. I fremskredne tilfælde kan ses trommestikfingre, cyanose og højresidigt hjertesvigt.
1.4.2 Paraklinik
Parakliniske værdier er sjældent af diagnostisk værdi for AA, men bør udføres i differentialdiag nostisk øjemed. Ved akut indsættende sygdom er de hyppigste fund leukocytose, forhøjet CRP, IgG, SR samt LDH.
Måling af specifikke IgG-antistoffer er kun indi ceret, hvis der er velbegrundet mistanke om, at en eksponering for relevante allergener kan have udløst AA. IgG-antistoffer er ikke ensbety dende med sygdom, men indikerer eksponering for det pågældende antigen. Mange asympto matiske personer kan have forhøjede værdier af specifikke IgG-antistoffer i blodet uden nogen sinde at udvikle AA. Der er fortsat en række ubesvarede spørgsmål vedrørende validering af test specifikke IgG
Flertallet af patienter har restriktiv lungefunkti onsnedsættelse med nedsat diffusion. Blandet restriktivt og obstruktivt mønster kan ses, mens et rent obstruktivt mønster er sjældent.
1.4.3 Billeddiagnostik
RU thorax kan ved non-fibrotisk AA være nor malt, men klassisk ses diffus øget lungetæthed med bilaterale diffuse mikronoduli basalt og perihilært. Ved fibrotisk AA ses bilaterale fibro seforandringer.
HRCT har en betydelig højere sensitivitet for AA end konventionel RU thorax, og bør altid udføres også hvor RU thorax er normalt og diagnosen AA fortsat mistænkes. Normal HRCT udelukker
dog ikke non-fibrotisk AA med kortvarig ekspo nering og resolution af inflammationen. Nye kriterier for HRCT inddeler fundene i typisk, forenelig med eller ubestemt for AA.
Non-fibrotisk AA
Den typiske HRCT viser diffus matglastegning med involvering af alle lungeafsnit med enkelte sparede lobuli, mosaikattenuering, centrilobulæ re mikronoduli og air trapping (figur 1). Foran dringerne er ofte reversible ved ophør af ekspo nering og med evt. steroidbehandling.
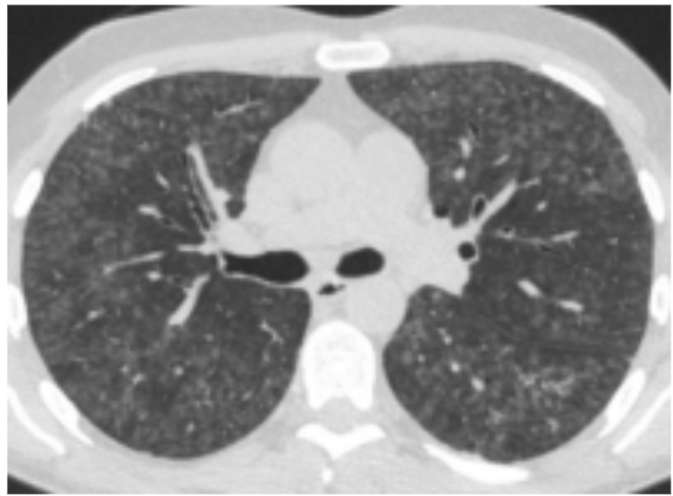
Figur 1. HRCT ved non-fibrotisk allergisk alveoli tis.
Fibrotisk AA
HRCT ved Fibrotisk AA inddeles ligeledes i ”ty pisk”, ”forenelig med” eller ”ubestemt” for fibro tisk AA. Ved den typiske fibrotiske AA ses retiku lering, traktionsbronkiektasier og evt. bikageteg ning i de mellemste og basale lungeafsnit. Sam tidig ses tegn på small airways disease med
centrilobulære noduli, mosaikattenuering og air trapping (figur 2). Fund af ”three-density-sign”, dvs. samtidig forekomst af matglastegning (øget attenuation), normalt lungevæv (normal attenua
tion) og mosaikattenuering (nedsat attenuation) er patognomonisk for fibrotisk AA. Tilstedevæ relse af fibrose er forbundet med en dårligere prognose.
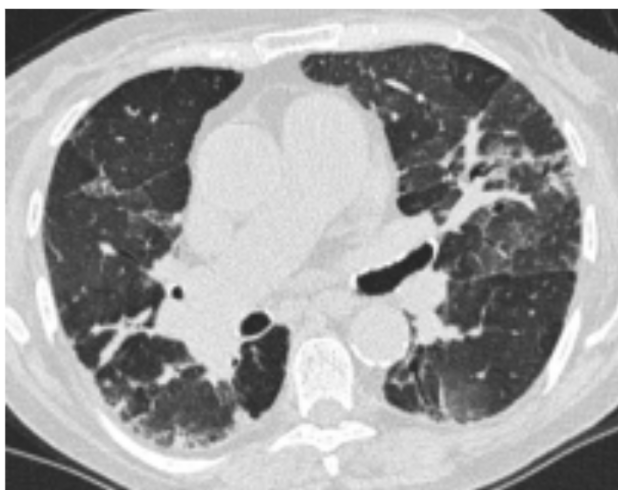
Figur 2. HRCT ved fibrotisk allergisk alveolitis.
1.4.3 Patologi
Ved bronkoalveolær lavage (BAL) findes typisk lymfocytær inflammation (lymfocytter 20-70%) og lav CD4/CD8 ratio ved non-fibrotisk AA, hvor imod der kan være normal BAL ved fibrotisk AA.
Man kan ofte opnå tilstrækkelig og repræsenta tiv histopatologi ved transbronkiale tang- eller cryobiopsier ved AA, men kirurgisk biopsi kan være nødvendige ved fibrotisk AA. Det histopa tologiske mønster inddeles i ”typisk”, ”forenelig med” og ”indeterminate”, hvor fund af bronkiolo centrisk inflammation, lymfocytær inflammation og løstformede non-nekrotiserende granulomer er klassiske fund ved den non-fibrotiske AA (fi gur 3). Der kan ses områder med organiserende pneumoni (OP) og non-specifik interstitiel pneumonitis (NSIP) foruden fibroseforandringer ved den fibrotiske AA. Fibrotiske forandringer viser sig typisk ved arkitektonisk distorsion, fi broblastfoci og bikagetegning, samt luftvejscen treret interstitiel fibrose.
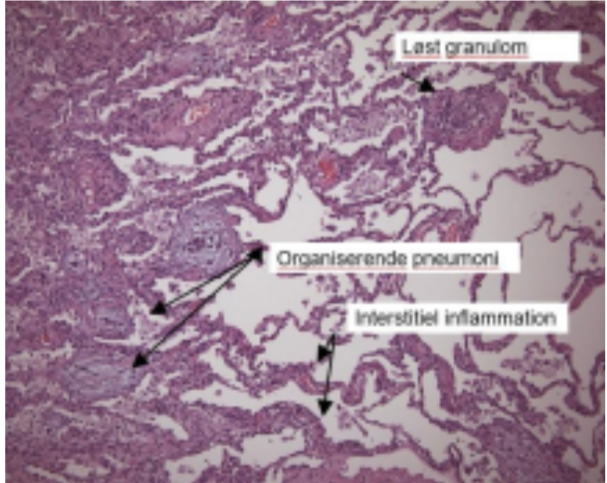
Figur 3: Typiske histologiske forandringer hos en pt. med non-fibrotisk AA (Patologisk institut; OUH)
Tilstedeværelse af fibrose er forbundet med en dårligere prognose. Jo mere fibrotisk det hist opatologiske mønster er, jo dårligere prognose (OP > NSIP > UIP).
1.5. DIAGNOSE
Hurtig og korrekt diagnose er vigtig idet syg dommen ved debut er potentielt reversibel, mens en udiagnosticeret og ubehandlet AA kan medføre varig lungeskade.
Grundlaget for at mistænke AA er det kliniske billede og radiologi som beskrevet i foregående afsnit.
Diagnosen baseres på grundig anamnese med fokus på eksponering både vedrørende er hvervs-, fritids- og beboelsesforhold herunder evt. fugle/dyr. Herudover afklares brug af læ gemidler. Henvisning til arbejds- og miljømedi cinsk afdeling anbefales ved mistanke om er
hvervseksponering eller ukendt agens. Det afgørende i diagnostikken er herefter at afklare om der er fund ved den objektive under søgelse, som styrker mistanken om AA og at HRCT er foreneligt med AA.
Mistanken om AA kan om muligt styrkes ved påvisning af specifikke IgG-antistoffer mod det mistænkte antigen. I 30-50% af tilfældene, og især ved fibrotisk AA, identificeres intet antigen eller specifikke IgG-antistoffer.
Især ved den non-fibrotiske form kan et diagno stisk værktøj være en mulig fjernelse af patien
ten fra det potentielt skadelige miljø (fx fjernelse fra arbejdspladsen eller hjemmet ved mistænkt eksponering) og observation af effekten af dette tiltag.
En undersøgelse (fx en byggeteknisk gennem gang) på det formodede ekspositionssted med påvisning af velkendt antigen kan sandsynliggø re en sammenhæng. Provokationstest kan udfø res som overvåget re-eksponering i patientens
miljø, fx på arbejdspladsen men benyttes sjæl dent i praksis pga. bekymring for mulig forvær ring af tilstanden.
Ved diagnostisk usikkerhed overfor anden in terstitiel lungesygdom er bronkoskopi med BAL, evt. TBB/TBCB, og i særlige tilfælde kirurgisk lungebiopsi indiceret.
Hvor mange undersøgelser af de nævnte, der skal gennemføres ved den enkelte patient af hænger af patientens risikoprofil og hvor sikker en diagnose, der ønskes (Se figur 4 nederst i retningslinjen).
Generelt bør der altid foreligge:
- Information om erkendt årsag/antigen 2. Udelukkelse af anden form for interstitiel lungesygdom
- Typiske fund på HRCT
Disse undersøgelser kan suppleres med: 1. BAL med lymfocytose (>30% for ikke el ler ex-rygere og > 20% for rygere); CD4/CD8 < 1.3
- Transbronkial eller kirurgisk lungebiopsi tydende på AA
- Evt. specifikke IgG antistoffer relevante for sygehistorien.
- Svind af symptomer ved fjernelse af pa tienten fra ekspositionen (eller evt. posi tiv provokationstest/re-eksposi-tion i pa tientens miljø).
1.6. DIFFERENTIALDIAGNOSE
Ved akut præsentation kan non-fibrotisk AA forveksles med influenza og pneumoni. Øvrige differentialdiagnoser omfatter Organic Toxic Dust Syndrome (OTDS) og Inhalation fever, hvor der ikke er en immunologisk reaktion.
Ved den længerevarende non-fibrotiske eller fibrotiske AA, hvor hovedsymptomerne er dyspnø, hoste og træthed kan sygdommen for veksles med KOL, astma eller andre interstitielle lungesygdomme som fx IPF, eller sarkoidose.
1.7. BEHANDLING
Tidlig diagnose og eliminering af antigen ekspo nering er de væsentligste elementer i behand lingen. Bortskaffelse af fugle, eller grundig un dersøgelse af boligen og arbejdspladsen med
efterfølgende sanering af evt. koloniserede byg ninger eller anlæg kan være en sufficient inter vention hos nogle patienter. Hvis antigenet ikke kan identificeres eller eliminering af eksponerin gen er umuligt at gennemføre i praksis, kan det blive nødvendigt at fjerne patienten fra det miljø, hvor eksponering forekommer. Dette kan have alvorlige sociale og økonomiske konsekvenser og bør overvejes nøje i samråd med arbejdsme-
diciner og de sociale myndigheder.
Ved non-fibrotisk AA er der evidens for, at be handling med steroid accelererer reduktion af symptomer, men uden at ændre den langsigtede prognose sammenlignet med placebo. Behand lingen bør derfor primært vælges til patienter med nedsat lungefunktion samt betydelige symptomer, som forringer livskvaliteten. Den optimale dosis og længde af behandlingen er uvis. Oftest startes med tbl. prednisolon (0,5-1 mg/kg dagligt) i 1-2 uger og herefter nedtrappes efter klinisk respons og lungefunktion samtidig med effektiv eliminering af det udløsende anti gen. Der foreslås en samlet behandlingsvarig hed på 3-12 måneder.
Ved fibrotisk AA gælder tilsvarende at eliminati on af det udløsende antigen har stor positiv be tydning for prognosen. Ved kombineret inflam mation og fibrose forsøges ofte kombinations behandling med prednisolon og/eller steroid besparende immunsuppression med azathioprin
eller mycophenolat mofetil, som vurderes at have ligeværdig effekt. Der foreligger ingen randomiserede undersøgelser.
Ved progression af symptomer, fald i lungefunk tion og tiltagende radiologiske forandringer trods standardbehandling (antigen elimination, im munomodulation) som udtryk for progressiv pulmonal fibrose (PPF) kan antifibrotisk behand ling med nintedanib (og ved uholdbare bivirknin ger pirfenidon) nedsætte progression af fibro sen.
Effekten af inhalationssteroid er ikke belyst, men kan forsøges evt. i kombination med LABA hos patienter med generende hoste og effekten re vurderes efter 2-3 måneder.
Patienter med nedsat lungefunktion (FVC<80% eller DLCO<40%), signifikant fald i lungefunktio nen (10% fald i FVC eller 15% fald i DLCO over 6 mdr.), hvilehypoksæmi eller pulmonal hyper tension skal vurderes mhp. lungetransplantation
såfremt der ikke er kontraindikationer herfor. Optimal palliation inkl. iltbehandling, lungereha bilitering og symptomlindrende medicin bør til bydes alle patienter.
Behandling af AA er en opgave for de højtspeci aliserede lungemedicinske afdelinger.
1.7. PROGNOSE
Risikofaktorer for dårlig prognose er alene belyst i retrospektive studier og omfatter bl.a. tilstede værelse af fibrose, manglende identifikation af antigen, demografi, og genetiske faktorer (tabel 2).
1.8. EFTERBEHANDLING OG KONTROL Valg af kontrol ved AA afhænger af klinisk præ sentation, intensitet og progression af sygdom men. Ideelt bør patienter med non-fibrotisk AA kontrolleres indtil helbredelse eller indtil stabilitet uden medicinsk behandling. Ved kontrol vurde res graden af respiratoriske symptomer, dyna misk spirometri og måling af diffusionskapacite ten samt evt. 6 minutters gangtest. Rutine kon
trol HRCT er ikke indiceret, men bør overvejes ved sygdomsprogression til at vurdere forholdet mellem reversible og irreversible forandringer. En praktisk tilgang er at afslutte patienten ved stabile forhold gennem 12 (-24) mdr.
Patienter med fibrotisk AA har ofte behov for langvarig, evt. livslang kontrol pga. behandling med immunmodulerende og/eller antifibrotisk behandling, risiko for kronisk respirationsinsuffi
ciens, pulmonal hypertension og øget mortalitet.
Tabel 2. Risikofaktorer for dårlig prognose Patient-relaterede
Alder
Mandligt køn
Genvarianter i telomerkomplekset
Samtidig forekomst af autoimmun sygdom
Eksponering
Uidentificeret eksponering
Langvarig eksponering
Højintens eksponering
Rygeanamnese
Kliniske
Lav lungefunktion (FVC, DLCO)
Desaturation ved aktivitet
Radiologisk fibrose (bikagetegning, UIP)
Histologisk fibrose
Pulmonal hypertension
1.9. REFERENCER
Raghu G et al. American Thoracic Society, Japanese Respiratory Society, and
Associacion Latinoamericana de Torax. Diagnosis of Hypersensitivity Pneumonitis in Adults: An Official ATS/JRS/ALAT Clinical Practice Guideline. Am J. Resp Crit Care Med. 2020.
Fernández Pérez ER, Travis WD, Lynch DA. Diagnosis and Evaluation of Hypersensitivity Pneumonitis. Chest 2021;160(2): e97-e156.
Silva CI, Churg A, Müller NL. Hypersen-sitivity pneumonitis: spectrum of high-resolution CT and pathologic findings. Am. J. Roentgenol. 2007;188(2): 334-44.
Kokkarinen JI, Tukiainen HO, Terho EO. Effect of corticosteroid treatment on the recovery of pulmonary function in farmer’s lung. Am. Rev. Respir. Dis. 1992;145(1): 3-5.
Morell F, Roger A, Reyes L, Cruz MJ, Murio C, Muñoz X. Bird fancier’s lung: a series of 86 pati ents. Medicine (Baltimore). 2008; 87(2): 110-30
Salisbury ML, Myers JL, Belloli EA. Diagnosis and Treatment of Fibrotic Hypersensitivity Pneumonia. Am J Respir Crit Care Med. 2017;196(6):690–699.
Riario Sforza GG, Marinou A. Hypersensitivity pneumonitis: a complex lung disease. Clin Mo lecular Allergy. 2017;15:6.
Morriset et al. Use of Mycophenolate Mofetil or
Azathioprine for the Management of Chronic
Hypersensitivity Pneumonitis. Chest 2017.
Flaherty et al. Nintedanib in Progressive Fibro
sing Interstitial Lung Diseases. N Engl J Med
2019; 31;381(18):1718-1727.
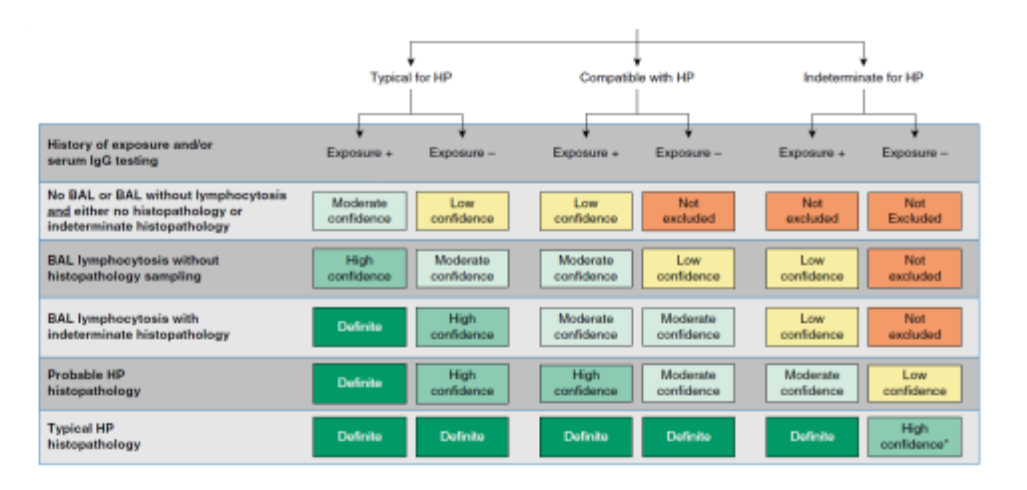 Figur 4. Diagnostisk algoritme for allergisk al
Figur 4. Diagnostisk algoritme for allergisk al
veolitis (Raghu et al. AJRCCM 2020).