
1.1. BAGGRUND
Interstitielle lungesygdomme (ILS) er en heterogen gruppe af lungesygdomme, som kan opstå sekundært til miljøpåvirkning, underliggende autoimmun sygdom eller af ukendte årsager. ILS kan udvikles som følge af inflammation med efterfølgende fibrosedannelse (arvævsdannelse) eller debutere med fibrose. Idiopatisk pulmonal fibrose (IPF) er den hyppigst forekommende arketype på fibrotisk ILS (se venligst DLS retningslinjen for ”Idiopatisk pulmonal fibrose”), men andre undertyper af ILS kan også præsentere sig med progressiv fibrose på HRCT af thorax i kombination med tiltagende symptomer, faldende lungefunktion og dårlig prognose, helt som IPF.
Frem til 2022 blev denne fænotype refereret til som progressiv fibrotisk ILS (PF-ILS). I februar
2022 godkendte Medicinrådet antifibrotisk behandling med nintedanib (Ofev ®) til patienter opfyldende kriterier for PF-ILS (se afsnit 1.7.). Siden maj 2022 har den internationalt accepterede definition for progressiv pulmonal fibrose (PPF) været anvendt, men kriterierne for opfyldelse af PPF er imidlertid endnu ikke godkendt af Medicinrådet som indikation for antifibrotisk behandling. Brugen af PF-ILS og PPF kan derfor skabe forvirring, men fælles for både PF-ILS og PPF er, at de hver især er defineret ved specifikke kriterier, og at der for begge definitioners vedkommende ikke refereres til én selvstændig sygdomsenhede, men til en fænotype som omfatter en heterogen gruppe af ILS, der inden for veldefinerede tidsrum fænotypisk præsenterer sig ved varierende grader af progressiv lungefibrose og inflammation (Figur 1).
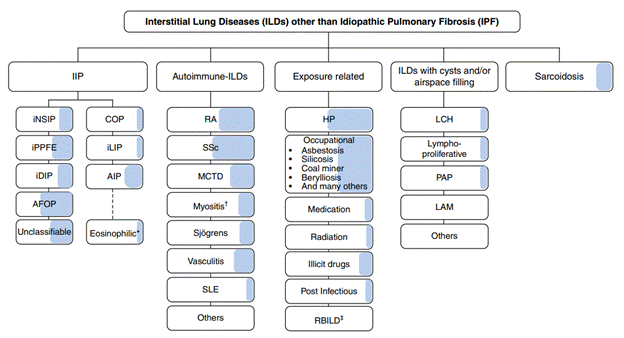
Figur 1: De lyseblå områder angiver den estimerede andel af patienter inden for en given ILS-subtype, som kan udvikle sig til manifest PPF (reference #8: Raghu G et al. Am J Respir Crit Care 2022(9);205:e18-e47).
1.2. DEFINITION
Progressionskriterierne for de 2 anvendte definitioner adskiller ved primært tre forhold: 1) Observationsperioden for progression af lungefibrose, 2) relative vs. absolutte fald i lungefunktionsværdier, og 3) inklusion af DLCO for PPF modsat for PF-ILS.
PF-ILS
For opfyldelse af definition for PF-ILS skal minimum 1 af flg. progressionskriterier skal være opfyldt inden for de sidste 24 måneder trods evt. standardbehandling (INBUILD kriterier):
- Relativt fald i FVC (% af forventet) ≥10 %
- Relativt fald i FVC (% af forventet) på 5 til <10 % OG forværret fibrose på HRCT
- Relativt fald i FVC (% af forventet) på 5 til <10 % OG forværrede symptomer*
- Forværrede symptomer* OG forværret fibrose på HRCT
*Anden årsag til forværrede respiratoriske symptomer skal udelukkes.
PPF
For opfyldelse af definition for PPF skal minimum 2 flg. progressionskriterier skal være opfyldt inden for de sidste 12 måneder trods evt. standardbehandling (ATS/ERS/JRS/ALAT kriterier):
- Forværrede symptomer*
- 1 af flg. lungefunktionskriterier:
- Absolut fald i FVC ≥ 5%
- Absolut fald i DLCO ≥ 10%
- 1 af flg. radiologiske kriterier på HRCT:
- Tiltagende grad og sværhedsgrad af traktionsbronkiektasier og traktoinsbronkiolektasier
- Nytilkomne matglasforandringer med traktionsbronkiektasier
- Nytilkommen retikulering
- Tiltagende grad af retikulering eller tiltagende grov retikulering
- Nytilkommen eller tiltagende grad af bikagetegning
- Tiltagende volumentab
*Anden årsag til forværrede respiratoriske symptomer skal udelukkes.
Uagtet opfyldelse af kriterier for PF-ILS eller PPF gælder det, at definitionerne ikke inkluderer idiopatisk pulmonal fibrose (IPF). I nærværende retningslinje anvendes generelt PPF, fraset når behandling omtales (afsnit 1.7.).
1.3. FOREKOMST.
Incidensen af PPF er svær at vurdere, men det skønnes, at ca. 20-30% med fibrotisk ILS vil udvikle sig til PPF. Dette antal forventes at stige pga. den forventede øgede opmærksomhed på mulig behandling (se senere).
1.4. ÆTIOLOGI
PPF dækker over flere ILS-typer og har således forskellig underliggende ætiologi for fibroseudvikling (Figur 1). De hyppigste typer er pulmonal manifestation af autoimmune sygdomme, hvor reumatoid artritis og sklerodermi har højest prævalens af associeret ILS, non-IPF idiopatiske interstitielle pneumonier (IIP), hypersensitivitetspneumonitis samt sarkoidose og pneumokonioser. Et fælles patofysiologisk træk er, at alle subtyper præsenterer sig med varierende grader af lungefibrose (Figur 1).
1.5. SYMPTOMER OG KLINISKE FUND
Som for andre ILS (se venligst DLS retningslinjen ”Interstitielle lungesygdomme”) er de mest almindelige symptomer dyspnø og hoste, som forværres over tid (måneder til år). Objektivt kan der ses clubbing i relation til enkelte PPF subtyper, men også hypoxæmi, der kan medføre pulmonal hypertension. Ved lungestetoskopisk vil man evt. kunne høre velcrokrepitation. Hos patienter med underliggende autoimmun gigtsygdom vil man ofte kunne identificere ekstrapulmonale symptomer fra bl.a. hud, led, slimhinder, muskler, øjne og tåre- og spytkirtler.
1.6. UDREDNING
PPF medfører gradvis forværring af respiratoriske symptomer, faldende lungefunktion og tiltagende fibrose på HRCT-skanning. Patienter med verificeret PPF vil derfor som udgangspunkt have et kontrolforløb på et af de højtspecialiserede ILS-centre og afklaring af underliggende ætiologi vil være kendt. I enkelte tilfælde kan en patient naturligvis debutere med subjektive og objektive fund forenelig med PPF, men ikke nødvendigvis opfylde kriterier for antifibrotisk behandling (se afsnit 1.6.2).
1.7. BEHANDLING
Den farmakologiske behandling baserer sig på:
- Antifibrotisk behandling med nintedanib (se senere)
- Behandling af evt. underliggende sygdom (f.eks. autoimmune gigtsygdomme) med bl.a. antiinflammatorisk behandling med glukokortikosteroider og immunsuppression. Evt. kombinationsbehandling med immunsuppression og antifibrotisk behandling besluttes individuelt, evt. ved MDT.
1.7.1. Nintedanib (Ofev®) og effektmål
Nintedanib (Ofev®) er en tyrosinkinasehæmmer, der hæmmer en række væksfaktorer, som fører proliferation, migration og differentiering af lungefibroblaster og videre udvikling af lungefibrose.
Mortaliteten af PPF korrelerer i høj grad med fald i FVC som følge af progression af lungefibrosen. Behandlingsmålet er derfor opbremsning af sygdomsudvikling med henblik på uforandret status eller reduceret progressionshastighed. I INBUILD studiet viste, at behandling med nintedanib sv.t. 150 mg x 2 dagligt igennem 52 uger kunne reducere sygdomsprogression målt ved et reduceret årligt FVC-fald på mellem 107-128 ml sammenlignet med placebogruppen. Studiet kunne ikke vise signifikante forskelle i dødelighed, livskvalitet eller tiden til exacerbation, men var heller ikke designet til at undersøge mortalitet.
1.7.2. Indikation for behandling med nintedanib
I henhold til Medicinrådets anbefaling kan patienter, der opfylder kriterier for PF-ILS, tilbydes behandling med nintedanib (se afsnit 1.2). For en selekteret mindre gruppe af patienter med progressiv fibrose, som har overvejende fibrotisk sygdom uden betydelig inflammation bedømt på HRCT +/- patologi er det oplagt, at nintedanib kan anvendes som førstelinjebehandling.
1.7.3. Dosering med nintedanib
Kapsel Ofev® findes i doser sv.t. 100 mg og 150 mg.
Opstartsdosering sv.t. kapsel Ofev®150 x 2 dagl., og ved bivirkninger (se afsnit 1.6.5) med mulighed for dosisreduktion til 100 mg x 2 dagl.
1.7.4. Indikation for behandlingsophør med nintedanib
Medicinrådet har besluttet, at der skal være stopkriterier for behandling med nintedanib. Behandlingen skal således seponeres hvis flg. kriterier opfyldes:
- Gennemført lungetransplantation
- Kronisk lungesvigt (behov for døgn-ilttilskud) og dårlig performansstatus 3 eller 4
- FVC-fald på samlet set >10% i løbet af ét år (målt ved 3 uafhængige målinger), trods stabilt indtag af nintedanib
1.7.5. Bivirkninger og håndtering ved nintedanib
Bivirkningerne er dosisafhængige. Kapsler skal indtages med mad, synkes hele med vand og må ikke tygges eller knuses. Målet med nintedanib behandling er, at flest mulige ptt. tager maksimal dosis pga. dosis-effekt sammenhængen. Dosis modificeres afhængig af bivirkninger (tilråd f.eks. indtag sammen med mad). Hvis nintedanib stoppes, så forsøg at genoptage behandlingen, evt. lidt langsommere og overvej altid, om bivirkningen er associeret til nintedanib eller evt. til noget andet.
GI-bivirkninger
De hyppigste bivirkninger er abdominalsmerter, diarré, kvalme, nedsat appetit, vægttab, opkastning og leverpåvirkning.
Håndtering af GI-bivirkninger
Diarré
- Instruér ptt. i, at diarré er en hyppig, men oftest mild bivirkning. Ved diarré påbegyndes loperamid og rehydrering.
- Stop evt. laksantia.
- Ved fortsatte symptomer dosisreduktion til 100 mg x 2, med efterfølgende stigning til den maksimalt tolererede dosis.
- Ved fortsatte symptomer trods dosisreduktion gøres 2 ugers behandlingspause, herefter genoptages behandlingen (100 mg x 2).
Kvalme
- Kvalme/opkastninger: Påbegynd anti-
- Ved fortsatte symptomer dosisreduktion til 100 mg x 2, med efterfølgende stigning til den maksimalt tolererede dosis.
- Ved fortsatte symptomer gøres 2 ugers behandlingspause, herefter genoptages behandlingen (100 mg x 2).
Leverpåvirkning
- Ved ALAT-forhøjelse > 3 gange øvre grænse for normalområdet skal man søge at udelukke andre årsager, herunder anden medicin og alkohol.
- Ved vedvarende ALAT forhøjelse reduceres dosis til 100 mg x 2 eller behandlingen pauseres indtil normale levertal.
- Behandlingen genoptages herefter med 100 mg x 2 med evt. senere dosisøgning.
- Ved ALAT > 3 gange øvre grænse for normalområdet og samtidige symptomer på nedsat leverfunktion seponeres nintedanib uden genoptagelse.
1.7.6. Kontraindikationer for behandling med nintedanib
For en selekteret gruppe patienter skal man være opmærksom på ordination af nintedanib. Det drejer sig om f.eks. patienter med:
- Svært nedsat leverfunktion
- Svært nedsat nyrefunktion (lav eGFR). Er patienten i dialyse ikke oplagt kontraindikation
- Allergi overfor jordnødder eller soja
- Abdominal kirurgi indenfor de sidste 4 uger
1.7.7. Anden opmærksomhed ved behandling med nintedanib
Patienter, som er bloddonorer, skal udmeldes af donorkorpset, når nintedanib startes. Visse tilstande kan betyde, at nintedanib skal pauseres eller seponeres. Det kan dreje sig om:
- Perforeret mavesæk eller tarm
- Elektiv operation, f.eks. aneurismeoperation
- Større sår (nintedanib kan potentielt hæmme sårhelingen)
1.7.8. Anden antifibrotisk behandling
Flere mindre studier har vist tilsvarende antifibrotisk effekt af pirfenidon hos patienter med PF-ILS og PPF, hvorfor pirfenidonbehandling anbefales såfremt nintedanib ikke tolereres.
1.7.9. Patientuddannelse
Brug tid på at instruere ptt. grundigt ved start på nintedanib. Gennemgå bivirkninger, forventet varighed samt giv information om f.eks. loperamid og rehydrering. På flere ILS-centre tilbydes denne patientkategori informationsmøder, hvor også behandling inkl. bivirkninger omtales.
1.8. KONTROL
Ved opstart af behandling med nintedanib
Der er national konsensus om, at der jf. mulige bivirkninger foretages blodprøvekontrol indeholdende, leverenzymer og væsketal initialt hver måned de første 6 måneder.
Igangværende behandling med nintedanib
Da PPF er kronisk tilstand, vil der være behov for langvarig opfølgning. Sygdommene præsenterer sig dog med varierende progressionsgrad, hvorfor der anbefales klinisk opfølgning jf. ”disease behaviour classification”, som er skitseret i DLS retningslinjen for ”Interstitielle lungesygdomme” (se venligst denne). Opfølgning bør indeholde blodprøver, spirometri og diffusionskapacitet, evt. 6 min. gangtest og HRCT thorax ved mistanke på forværring eller som led i differentialdiagnostik. Blodprøver bør kontrollers hver 3. måned, og på baggrund af sygdomssværhedsgrad og sygdomsaktivitet anbefales klinisk kontrol ca. hver 6.-12. måned
1.9. PROGNOSE
Der eksisterer overlap imellem de forskellige studiers definitioner af progressiv lungefibrose, som kan vanskeliggøre regelrette epidemiologiske mål herfor. De patogenetiske mekanismer, det kliniske og radiologiske sygdomsbillede samt prognose er sammenlignelige mellem PPF og IPF, og PPF er således forbundet med nedsat funktionsniveau, forværret livskvalitet og tidlig død trods immunmodulerende og antifibrotisk behandling. Patienternes prognose er afhængig af lungefibrosens omfang, hvorvidt usual interstitial pneumonia (UIP) mønster er til stede, hvor hurtigt lungefunktion falder og frekvensen af akutte eksacerbationer. Herudover er det påvist, at høj alder, mandligt køn, GERD og lav FVC samt DLCO er risikofaktorer for død. Gennemsnitsoverlevelsen fra diagnosetidspunkt afhænger af underliggende ILS-subtype, men ved manifest UIP estimeres en overlevelse at være 2-10 år fra diagnosetidspunkt afhængigt af underliggende ILS subtype.
For selekterede patienter bør man på et tidligst tidspunkt overveje mulighed for lungetransplantation (se venligst DLS retningslinjen for ”Lungetransplantation”), og ved persisterende sygdomsprogression og manglende effekt af antifibrotisk behandling anbefales ligeledes henvisning til palliative teams.
1.10. REFERENCER
- Dansk Lungemedicinsk Selskab. Retningslinjer – Interstitielle lungesygdomme (ILS). 2020; Tilgængelig fra: https://lungemedicin.dk/interstitielle-lungesygdomme-ils/
- Dansk Lungemedicinsk Selskab. Retningslinjer – Idiopatisk pulmonal fibrose (IPF). 2020; Tilgængelig fra: https://lungemedicin.dk/idiopatisk-pulmonal-fibrose-ipf/
- Medicinrådets protokol for vurdering vedr. nintedanib til PF-ILS-vers. 1.0 (medicinraadet.dk)
- Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y, et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N Engl J Med. 2019;381(18):1718–27.
- Distler O, Highland KB, Gahlemann M, Azuma A, Fischer A, Mayes MD, et al. Nintedanib for Systemic Sclerosis–Associated Interstitial Lung Disease. N Engl J Med. 2019;380(26):2518–28.
- Wijsenbeek M, Cottin V. Spectrum of Fibrotic Lung Diseases. Drazen JM, red. N Engl J Med. 2020;383(10):958–68.
- Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2018;198(5):e44–68.
- Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2022;205(9):e18-e47.
- Wells AU, Brown KK, Flaherty KR, Kolb M, Thannickal VJ. What’s in a name? That which we call IPF, by any other name would act the same. Eur Respir J. 2018;51(5):1800692.
- Cottin V, Wollin L, Fischer A, Quaresma M, Stowasser S, Harari S. Fibrosing interstitial lung diseases: knowns and unknowns. Eur Respir Rev. 2019;28(151):180100.
- George PM, Spagnolo P, Kreuter M, Altinisik G, Bonifazi M, Martinez FJ, et al. Progressive fibrosing interstitial lung disease: clinical uncertainties, consensus recommendations, and research priorities. Lancet Respir Med. 2020;8(9):925–34.
- Brown KK, Martinez FJ, Walsh SLF, Thannickal VJ, Prasse A, Schlenker-Herceg R, et al. The natural history of progressive fibrosing interstitial lung diseases. Eur Respir J. 2020;55(6):2000085.
- Wollin L, Distler JHW, Redente EF, Riches DWH, Stowasser S, Schlenker-Herceg R, et al. Potential of nintedanib in treatment of progressive fibrosing interstitial lung diseases. Eur Respir J. 2019;54(3):1900161.
- Dansk Lungemedicinsk Selskab. Retningslinjer – Lungetransplantation. 2023; https://lungemedicin.dk/lungetransplantation/
- Hambly N, Farooqi MM, Dvorkin-Gheva A, Donohoe K, Garlick K, et al. Prevalence and characteristics of progressive interstitial lung disease in a prospective registry. Eur Respir J 2022;60:2102571.
- Nasser M, Larrieu S, Boussel L, Si-Mohamed S, Bazin F, et al. Estimates of epidemiology, mortality and disease burden associated with progressive fibrosing interstitial lung disease in France (the PROGRESS study). Respir Res 2021;21:162.
- Khor YH, Farooqi M, Hambly N, Kolb M, Ryersen CJ. Patient characteristics and survival for progressive pulmonary fibrosis using different definitions. Am J Respir Crit Care Med 2023;207:102-105.